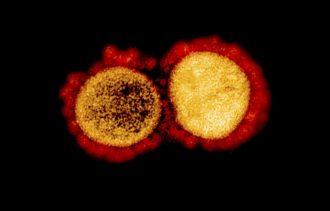
1 – Qu’est-ce qu’une mutation ?
L’immense influence de la science-fiction sur l’imaginaire collectif connote le terme « mutation » d’une aura spectaculaire, qui le déforme dramatiquement, en lui conférant de nouvelles capacités extraordinaires. En réalité, une mutation est infiniment moins spectaculaire et infiniment plus complexe à caractériser. Il faut revenir à ce qui mute, c’est-à-dire le génome.
Le génome est le « plan » de chaque entité biologique, des organismes pluricellulaires animaux aux virus. Constitué d’ADN dans la plupart des entités biologiques (dont les animaux et l’homme), il est composé d’ARN chez certains virus (c’est notamment le cas du Sars-Cov-2). Dans tous les cas, ce génome à pour fonction principale (même si l’on découvre jour après jour que, comme à peu près tout en science, c’est bien plus compliqué que cela) de permettre in fine la production de protéines qui vont constituer les cellules, soit directement, soit indirectement en catalysant les réactions chimiques permettant la production de ces composant et leur assemblage. Le cheminement considéré comme « classique » (comme beaucoup de choses en sciences, c’est parce qu’il a été découvert en premier) et appelé ni plus ni moins « dogme central de la biologie moléculaire » dit que l’ADN, situé dans le noyau, sert de presse d’imprimerie permettant de produire des molécules d’ARN messager (oui, celles du vaccin) qui sont en quelque sorte des photocopies d’une partie de l’ADN, qui pourront sortir du noyau et servir à leur tour de recettes à partir desquelles seront faites les protéines.
En digressant brièvement on peut d’ailleurs voir ici pourquoi un vaccin contenant de l’ARN messager ne serait pas en mesure de modifier le génome, et l’intérêt d’utiliser de l’ARN messager plutôt que des protéines. Tout d’abord l’ARN messager intervient en aval du génome à ADN contenu dans nos cellules, il n’a pas la capacité d’influencer le contenu de l’ADN dont il n’est censé être qu’une photocopie partielle et transitoire, dégradée en quelques minutes à quelques heures par nos cellules. Mais un des intérêts d’employer de l’ARN messager contenant le plan de protéines du virus plutôt que directement des protéines du virus vient dans l’effet d’amplification : en remontant d’un cran dans la chaîne de production on démultiplie la quantité de protéines produites au final à partir d’une même dose, une seule molécule d’ARN messager permettra de produire au moins plus d’une dizaine de protéines, or ce sont ces protéines qui doivent déclencher une réaction du système immunitaire et le pousser à produire une réponse dirigée contre elle qui sera également efficace contre le « vrai virus ».
Revenons aux mutations.
Une définition très rapide et simpliste d’une mutation serait de dire qu’ au moins un « caractère » de la séquence génomique, ADN dans la plupart des cas et ARN dans le cas de certains virus, est modifié. Là où les choses se compliquent, c’est lorsqu’il s’agit d’établir par rapport à quoi ce caractère est modifié. Chez la plupart des espèces il existe une séquence dite « consensus » qui contient au niveau de chaque position du génome le caractère statistiquement le plus prévalent dans l’espèce. Une mutation consisterait alors en la présence d’un autre caractère que celui-ci à cet emplacement. Oui mais… chez la plupart des espèces, les individus sont tous, à des degrés divers, différents, et comportent tous, au moins à certains endroits, des caractères qui divergent de la séquence dite « consensus ».
Chez la plupart des espèces il existe une séquence dite « consensus » qui contient au niveau de chaque position du génome le caractère statistiquement le plus prévalent dans l’espèce.
Xavier Olessa-Daragon
Serions-nous alors tous, à des degrés divers, des mutants ? Oui. Mais l’idée derrière le terme de mutation est plus l’apparition d’un nouveau caractère. Dans le cas de l’espèce humaine et, par exemple, de la couleur des yeux, il existe un certain nombre de variations des séquences des gènes influant sur la couleur des yeux répartis dans la population, toutes issues au départ de mutations, mais s’étant depuis suffisamment établies dans la population pour qu’il devienne relativement complexe de définir une « couleur d’yeux consensus ». Par mutation on va donc généralement plutôt faire référence à l’apparition d’une nouvelle version du génome d’une espèce.
2 – Comment les mutations apparaissent-elles ?
Même si elles jouissent de droits inaliénables tels que la vie, la liberté, et la recherche du bonheur, les entités biologiques ont en règle générale pour objectif principal leur propre réplication. Cette réplication passe ainsi bien sûr d’abord et avant tout par une réplication du génome. Sans entrer dans des détails trop techniques, disons que la machinerie qui opère cette réplication possède un taux d’erreur non nul, et qu’ainsi la réplication est rarement, voire jamais, parfaite, et que chaque processus de réplication va ainsi comporter en règle générale au moins une mutation sur les quelques milliers à quelques milliards de caractères du génome répliqué. Dans le cas de l’espèce humaine, ces mutations peuvent intervenir dans beaucoup de cas de figure que l’on peut simplifier en deux cadres, une réplication cellulaire, et une réplication à l’échelle d’un organisme.
Les cellules d’un individu se renouvelant, des mutations peuvent apparaître par exemple lorsqu’une cellule souche se divise pour produire une cellule qui a été endommagée ou qui est « arrivée à expiration », c’est entre autres par ce biais que peuvent apparaître les cancers. Le second cadre est celui de la reproduction, lors du processus de création des gamètes qui en se combinant donneront un nouvel organisme. Dans ce contexte, qui est celui où ces « mutations » seront transmises à la descendance, il est infiniment complexe de parler réellement de mutations dans la mesure où la nature elle-même a prévu des moyens d’augmenter significativement les recombinaisons, mutations et autres moyens d’augmenter les différences entre le génome contenu dans les gamètes et celui de leur géniteur, et ce afin d’augmenter le pool de diversité génétique. Car sans entrer dans trop de détails, disons que c’est au sein de ce pool de diversité génétique apparue lors de la production de gamètes que l’évolution va pouvoir sélectionner les « mutations » qui confèrent le plus d’avantages au sein d’un environnement donné.
On commence à entrevoir la double complexité derrière le terme de mutation : la référence par rapport à laquelle on considère la mutation (« nous sommes tous le mutant de quelqu’un »), et le fait qu’il s’agit d’une source d’innovations, pouvant ensuite être sélectionnées ou non par l’évolution. Dans le cas des virus, la réplication du génome est permanente. Les virus ont pour objectif premier d’entrer dans les cellules au sein desquels ils le peuvent (ce tropisme étant notamment guidé par les protéines de surface du virus, dans le cas du Sars Cov 2 la fameuse protéine S qui en interagissant avec la protéine de surface ACE 2 d’un certain nombre de nos cellules, notamment pulmonaires, va pouvoir y entrer), puis comme objectif second de détourner à leur profit toute la machinerie de ces cellules pour la rediriger vers un seul et unique but : la réplication du virus, et la production d’un maximum de copies du virus, avant d’arriver au dernier objectif principal, faire sortir toutes ces copies du virus de la cellule pour qu’elles puissent à leur tour pénétrer dans d’autres cellules et ainsi de suite. C’est le cycle de la vie.
Le virus se reproduisant en permanence, il mute infiniment plus vite que nous. Là où l’espèce humaine met au grand minimum une centaine et, le plus souvent, au moins des milliers d’années pour que certaines mutations se répandent, les virus peuvent, comme on le voit en ce moment, évoluer en quelques mois. Le rythme effréné de reproduction du virus sera donc la première raison de sa mutation rapide. Là ou un être humain, peut, dans les cas les plus extrêmes se reproduire une centaine de fois en une centaine d’années maximum (pour le moment) d’existence, ce chiffre est amplement battu au sein d’une seule cellule d’une seule personne infectée où au moins des milliers de copies du virus seront produites.
On commence à entrevoir la double complexité derrière le terme de mutation : la référence par rapport à laquelle on considère la mutation (« nous sommes tous le mutant de quelqu’un »), et le fait qu’il s’agit d’une source d’innovations, pouvant ensuite être sélectionnées ou non par l’évolution. Dans le cas des virus, la réplication du génome est permanente.
Xavier Olessa-Daragon
La seconde raison majeure de la mutation rapide de certains virus, a à voir avec la machinerie utilisée pour la réplication. La machinerie utilisée par l’espèce humaine a été perfectionnée au cours de l’évolution pour, dans le contexte de la réplication cellulaire au sein d’un individu, faire le moins d’erreurs possibles. Un grand nombre de mécanismes prévus spécifiquement pour détecter et corriger les erreurs existent, et on estime ainsi que la machinerie de réplication humaine fait environ une erreur de réplication pour 1 milliard de caractères répliqués. Le génome humain faisant environ 3 milliards de caractères, cela fait environ 1 à 3 mutations par réplications, l’écrasante majorité du temps sans conséquences.
La machinerie utilisée par les virus est très variable, parfois il s’agit essentiellement uniquement de la machinerie cellulaire humaine (notamment dans le cas des virus à ADN), et parfois elle est nettement, voire très nettement moins précise. L’exemple le plus extrême est probablement le VIH, dont la machinerie particulièrement peu fidèle au template qu’elle lit fait environ une erreur tous les dix mille caractères répliqués, soit cent mille fois plus d’erreurs que la machinerie humaine. Le Sars-Cov 2 utilise pour une large part sa propre machinerie pour se répliquer. Il est à ce stade très difficile d’avoir des chiffres précis quant au taux d’erreur mais il semble que sa machinerie soit nettement plus précise que celle du VIH, elle posséderait ainsi notamment des mécanismes de relecture, mais elle serait tout de même nettement moins précise que celle de l’espèce humaine.
Quand on multiplie le nombre de cellules infectées en moyenne chez une personne positive (au moins plusieurs milliers voir dizaines de milliers) par le nombre de personnes infectées (plusieurs millions) on entrevoit ainsi le nombre d’erreurs de réplications qui ont déjà été effectuées depuis le début de l’épidémie.
3 – Comment les étudie-t-on ?
Et plus que de les entrevoir, on peut justement les voir. Aujourd’hui tout le monde comprend à peu prêt ce qu’est une PCR : un moyen de savoir si l’on a le Covid-19 ou non. Sans rien remettre en cause et sans lancer aucun débat sur le sujet, on peut établir qu’il existait deux grandes stratégies techniques de dépistage de l’épidémie : la PCR, globalement plus rapide et techniquement plus facile à mettre en œuvre, mais qui offre un apport d’information restreint, et le séquençage massif et quasi systématique, plus long, plus cher, techniquement et logistiquement bien plus complexe à déployer, et offrant un flot d’information très riche mais bien plus complexe à analyser.
Sans entrer dans les détails techniques, la PCR consiste à utiliser une sonde correspondant à une région que l’on considère comme très peu mutée et mutagène du génome du virus pour venir voir si une séquence génomique correspondant à cette séquence est présente dans l’échantillon. On obtient une information qualitative grossière (la région génomique correspondant à la sonde employée est ou non présente, et avec elle le reste du génome du virus et donc le virus lui-même), et une information quantitative même si elle est la plupart du temps peu exploitée. En effet la technique employée est une RTqPCR — q pour quantitatif — la technique estimant la quantité de matériel génomique testé présent dans l’échantillon, et au-dessus d’un certain seuil excluant toute erreur ou toute détection d’une séquence quelque peu similaire mais n’ayant rien à voir avec le virus, déclarant que la personne est bien infectée par le virus avec une probabilité satisfaisante (en général autour de 95 %).
Mais cette technique ne permet pas d’en savoir plus sur la séquence précise du virus présent chez la personne, ni même si un virus trop muté pour être détecté par la sonde employée est présent, même si cette dernière hypothèse semblait jusqu’ici extrêmement peu probable. Jusqu’ici. De l’autre côté, le séquençage consiste comme son nom l’indique à séquencer, et donc à obtenir la séquence génomique de l’ensemble du matériel génétique contenu dans un échantillon, puis à voir si l’on trouve au sein de toutes ces séquences, celles du virus. La technique est non seulement plus coûteuse, et nécessite du matériel plus rare et plus cher, mais l’interprétation des données obtenues est infiniment plus complexe.
Là ou une formation express permet à beaucoup de gens de lire les résultats d’une RT-qPCR de dépistage, et de savoir si la différence entre le nombre de cycles d’amplification requis pour atteindre une quantité définie au départ de matériel génétique au sein de l’échantillon et au sein d’un échantillon d’eau servant de témoin négatif (dit ∆ Ct, pour Delta Cycle Treshold) est supérieure à un seuil fixé au départ, (par exemple 25 ou 30) et donc si la personne est positive, ou si elle est inférieure, et donc si la personne est négative, le traitement mathématique, bio-informatique et statistique requis pour interpréter des données de séquençage est considérable. Séquencer tous les échantillons de dépistage aurait ainsi offert une mine d’informations précieuses permettant de suivre presque en temps réel l’apparition de mutations dans la séquence du virus, mais probablement posé des défis logistiques considérables risquant de ralentir significativement les efforts de dépistage.
On a ainsi choisi dans presque tous les pays d’utiliser la PCR comme test de diagnostic courant, et de séquencer régulièrement un certain nombre d’échantillons pour suivre l’évolution de la séquence du virus. C’est ainsi que les scientifiques sud-africains observant l’apparition d’une « nouvelle souche » ont alertés leurs collègues, qui ont dans le monde entier augmenté rapidement et massivement leurs opérations de séquençage et ont ainsi découvert cette « nouvelle souche » au Royaume-Uni et maintenant en France et dans une trentaine d’autres pays. L’alerte sud-africaine a entraîné une démultiplication des opérations de « séquençovigilance » qui ont permis de réaliser l’ampleur de la diffusion des « nouvelles souches ».
4 – Une mutation, et ensuite ?
La plupart du temps, rien. La grande, voir très grande majorité des changements d’un caractère dans la séquence génomique n’ont aucun effet. Là où les choses se compliquent, c’est que certaines ont un effet « avantageux » (du point de vue de l’entité biologique, ici du virus), mais d’autres auront un effet « délétère ».
Qui fera le tri ? L’évolution, ni plus ni moins. Les virus porteurs de mutations « avantageuses » vont pouvoir par exemple se répliquer plus vite, et échapper mieux au système immunitaire, devenant petit à petit plus nombreux que ceux qui n’ont pas ces mutations qui vont eux se répliquer plus lentement et être plus neutralisés, on dit que la mutation se répand, parfois jusqu’à devenir présente dans presque toutes les souches du virus en circulation, devenant ainsi le « nouveau consensus » (quelqu’un observant la population à cet instant là considérerait alors l’absence de cette mutation devenue ultra majoritaire comme la mutation, relativisant là encore le concept de mutation). À l’inverse les virus porteurs de mutations « délétères », ralentissant l’efficacité du virus ou favorisant sa neutralisation par le système immunitaire, vont se répliquer moins vite, être plus facilement neutralisés, et la mutation va avoir tendance à disparaître. C’est là tout le principe de la sélection naturelle de Darwin.
De manière assez incroyable, certaines mutations sont « si avantageuses », qu’elles sont « apparues plusieurs fois », signifiant qu’elles sont apparues aléatoirement plusieurs fois à la suite d’erreurs de réplication, mais ont surtout été sélectionnées plusieurs fois tant elles sont avantageuses.
En digressant on peut d’ailleurs noter que le développement des premiers vaccins reposait précisément sur le fait de faire acquérir aux souches virales des mutations « avantageuses » pour leur réplication dans des cellules autres qu’humaines (de lapin pour le vaccin de la rage de Pasteur et de pomme de terre pour le BCG contre la tuberculose) mais « délétères » pour leur réplications au sein des cellules humaines (relativisant là encore la notion d’ « avantageux » ou « délétère »). Les nouvelles souches virales ayant accumulées ces mutations devenaient incapable de se répliquer assez efficacement dans des cellules humaines pour provoquer la maladie, mais restaient assez proches de la souche originale pour provoquer une réponse immunitaire qui serait ensuite efficace contre le « vrai » virus.
Concrètement, dans le cas du Sars-Cov-2, on peut évoquer trois principaux risques liés à l’apparition de mutations. Le premier est le fait qu’il s’agisse de mutations « avantageuses » pour le virus, et le rendant plus contagieux (ce qui semble être le cas), plus dangereux et plus mortel. Le second, tout aussi préoccupant, et qui préoccupe d’ailleurs plutôt plus les scientifiques et les autorités sanitaires, et le fait que le virus « change d’apparence » au point que les réponses immunitaires montées contre « l’ancienne souche » ne soient plus efficaces contre celle-ci. Ces risques s’appuient notamment sur l’apparition de mutations sur la fameuses « protéine S », S pour spicule, la protéine de surface du virus et qui est la cible utilisée par la plupart des vaccins actuellement déployés. Une nouvelle souche « trop différente » pourrait alors potentiellement faire chuter l’efficacité des vaccins, sans même parler des anticorps de ceux ayant déjà eu le Covid-19…
À ce titre, notons que les mutations sont le plus souvent nomenclaturées en indiquant d’abord la lettre correspondant à l’acide aminé (les briques LEGO constituant les protéines, au nombre d’environ 22) considéré jusqu’alors comme consensus, puis le numéro correspondant à l’emplacement de la mutation, et enfin l’acide aminé apparu suite à la mutation. Par exemple, N501Y, signifie que le 501e acide aminé consensus de la protéine était jusqu’alors N soit l’asparagine, et que la mutation le voit remplacé par Y soit la tyrosine. L’apparition de mutations au sein de la protéine S est donc potentiellement très préoccupante. Potentiellement parce qu’il est encore trop tôt pour connaître avec suffisamment de précision les conséquences de telles mutations. Le moyen principal par lequel une ou des mutations pourraient changer dramatiquement « l’aspect » du virus et notamment de sa protéine S passerait par la modification du repliement et de la structure tridimensionnelle. Or le repliement et la structure tridimensionnelle des protéines, et l’impact de mutations dessus est un champ incroyablement complexe, forçant sans cesse les chercheurs à repousser les limites de ce qu’ils sont capables de faire en termes de modélisation et de puissance de calcul informatique. De là à être capable de modéliser et simuler informatiquement avec précision les interactions entre des anticorps dirigés contre « l’ancienne » protéine S, et la nouvelle protéine S… Il est raisonnable de penser que nous aurons colonisé Mars avant d’être capables de le faire. Quand bien même on pourrait donc suivre en temps réel l’apparition de mutations, et évaluer leur potentiel de nuisance en repérant où elles se situent, et le degré de différence entre « le nouveau » et « l’ancien » acide aminé, on est incapable de savoir précisément quelles en seront les conséquences sur la contagiosité, la mortalité, ou la structure du virus en temps réel. Le troisième risque potentiel, tout aussi préoccupant, est l’augmentation du taux de faux négatifs des méthodes de dépistages qui ne seraient plus en mesure de détecter ces « nouvelles souches ». Nous serions en mesure de corriger le tir et de développer de nouvelles sondes de tests PCR capables de les détecter, mais le retard et la complexification considérables que cela entraînerait constitueraient déjà en soi un énorme problème.
De là à être capable de modéliser et simuler informatiquement avec précision les interactions entre des anticorps dirigés contre « l’ancienne » protéine S, et la nouvelle protéine S… Il est raisonnable de penser que nous aurons colonisé Mars avant d’être capables de le faire.
XAVIER OLESSA-DARAGON
Notons qu’émerge ici une question aussi intéressante que complexe : à partir de combien de mutations parle-t-on de « nouvelle » souche ? À partir de combien de mutations les anticorps et les tests diagnostiques deviennent-ils moins efficaces ? Plus du tout efficaces ? On ne sait pas répondre aux deux dernières questions. Pour ce qui est de la première, on peut tenter d’y répondre en introduisant l’idée de « parenté dynamique », à savoir que le virus évolue et change en permanence, qu’au sens propre du termes de « nouvelles souches » apparaissent de façon continue et que l’idée de « nouvelle souche » marque en réalité dans une large mesure une « rupture avec accumulation rapide de mutations » entre deux souches apparentées. Là où on estime que le virus accumulerait environ 1 à 2 mutations par mois, la « nouvelle souche britannique » aurait entre 14 et 22 mutations par rapport à son « ancêtre le plus proche » (la souche préalablement connue ayant la séquence la plus proche). Sans rentrer dans les détails, un des amendements postérieurs majeurs à la théorie de l’évolution de Darwin postule que l’évolution n’est pas un processus continu, mais qu’il comprend des phases lentes et des phases brutales d’évolution rapide. Ces nouvelles souches pourraient être dues à une ou plusieurs de ces phases.
5 – Quelles conséquences de ces « nouvelles souches » sur la gestion de l’épidémie ?
En termes politiques, on peut raisonnablement penser qu’elles vont contribuer à rendre encore plus explosive la question de la vaccination, notamment dans des pays avec une population aussi défiante sur la question que la France. La voix, déjà forte, des soignants épuisés qui poussent pour une vaccination plus contraignante, pourrait bientôt être rejointe par celle des scientifiques et des chercheurs expliquant que si l’on attend trop et qu’on ne vaccine pas dès maintenant tous ceux qui peuvent l’être, on laisserait potentiellement se développer de nouvelles souches en mesure de contaminer les vaccinés, et tout ceux qui l’ont déjà été…
En termes « techniques », cela signifie, comme c’est déjà le cas depuis l’alerte sud-africaine, qu’il faut multiplier les opérations de « séquençovigilance » afin de suivre l’évolution du génome du virus, et potentiellement adapter les sondes utilisées dans les tests PCR et dans les tests détectant la présence d’anticorps. A ce stade et sans informations précises laissant entendre que les tests de dépistage ne parviendraient pas à détecter ces « nouvelles souches », on en est pas encore à devoir significativement ajuster ces tests. Mais cela pourrait venir. Et potentiellement toucher également les vaccins…
La voix, déjà forte, des soignants épuisés qui poussent pour une vaccination plus contraignante, pourrait bientôt être rejointe par celle des scientifiques et des chercheurs expliquant que si l’on attend trop et qu’on ne vaccine pas dès maintenant tous ceux qui peuvent l’être, on laisserait potentiellement se développer de nouvelles souches en mesure de contaminer les vaccinés, et tout ceux qui l’ont déjà été…
Xavier Olessa-Daragon
6 – Peut-on anticiper les mutations ?
Pas vraiment. On sait que certaines régions du virus vont avoir plus tendance à muter que d’autres, parce qu’il existe certaines régions ou la moindre mutation aura des conséquences dramatiques sur la capacité du virus à entrer dans les cellules ou à se répliquer. On dit qu’il s‘agit de régions qui « tolèrent très peu de mutations » et ce sont généralement ces régions qui sont utilisées comme sondes lors des tests PCR. On sait à l’inverse que d’autres régions auront plus tendance à muter car des mutations pourraient conférer un avantage significatif au virus, il s’agit notamment des régions de surface qui vont aider le virus à entrer dans les cellules ou à se camoufler du système immunitaire. On sait enfin que plus le virus circule, plus il se réplique, plus il y aura de mutations, et donc plus la probabilité est grande que finissent par apparaître des mutations qui changent significativement son aspect. Mais l’on n’est pas réellement en mesure d’anticiper et de savoir quelles mutations vont apparaître et quel sera leur effet. Là encore la puissance de modélisation et de calcul informatique requis seraient titanesques.
7 – Concrètement que sont ces « nouvelles souches » ?
D614G, exemple d’un statu quo en réécriture perpétuelle
La « mutation » D614G concerne la protéine de surface du virus, la fameuse « Spike Protein », largement décisive en ce qui concerne la capacité du virus à pénétrer dans les cellules. Apparue en Europe en février, elle conférerait un tel avantage dans la capacité à pénétrer dans les cellules et à se répliquer du virus, que la sélection positive aurait finalement fait de la Glycine (G) l’acide aminé « consensus » à la position 614. Grâce à la réplication effrénée du virus générant un pool de variants, et à la pression de sélection faisant parmi eux émerger « les plus efficients », de « mutation » à « consensus » en quelques mois.
La « Souche Britannique » aussi nommée B.1.1.7 ou « VUI – 202012/01 » (Variant Under Investigation en Décembre 2020 numéro 1 ou encore 20B/501Y.V1)
La « souche britannique », B.1.1.7 serait apparue au Royaume-Uni en septembre 2020. Cette souche comprend donc entre 14 et 22 mutations spécifiques (selon le référentiel auquel on la compare — encore une fois on est tous le mutant de quelqu’un, mais c’est quoi qu’il en soit énorme), dont un certain nombre sont déjà caractérisées et pour lesquelles on aurait déjà des hypothèses concernant leurs conséquences.
La mutation N501Y, serait elle aussi située sur la protéine de surface du virus, la fameuse « Spike Protein » (parfois abrégée en protéine S), et plus précisément sur le RDB (Receptor Binding Domain) la partie du virus se liant à son récepteur sur les cellules humaines pour y entrer, ACE2, et augmenterait l’affinité du RBD pour ACE2, facilitant donc la capacité du virus à entrer dans les cellules. Même si encore une fois jusqu’ici on a pas d’informations indiquant que ces nouveaux variants échapperaient à l’immunité créée par les différents vaccins actuellement déployés, il est important de rappeler que ces vaccins utilisent pour la plupart précisément cette protéine S comme cible contre laquelle ils provoquent une réaction immunitaire.
La mutation P681H est proche d’un « site de clivage » par la furine 1. Sans rentrer dans les détails, un certain nombre de virus sont d’abords produits sous une forme nécessitant un certain nombre de modifications avant d’être opérationnelle. Parmi ces modifications se trouve souvent la coupure par des enzymes telles que la furine. Une mutation proche d’un site de clivage telle que celui-ci peut ainsi avoir un fort impact biologique, par exemple en modifiant la structure du virus et en facilitant l’accès de l’enzyme à la séquence qu’elle doit couper, augmentant l’efficacité de ce processus de coupure, et donc l’efficacité de réplication du virus.
Les mutations N501Y et P681H auraient été préalablement observées, mais jusqu’ici jamais ensemble au sein de la même souche.
La délétion des acides aminés 69 et 70, qui permettrait de supprimer une partie du virus bien reconnue par le système immunitaire, et lui permettrait ainsi de mieux y échapper. On voit encore ici comment la pression de sélection joue, les virus avec cette délétion seraient moins bien neutralisés par le système immunitaire que ceux ne l’ayant pas, et donc in fine plus nombreux à se reproduire, en les remplaçant peu à peu.
La mutations Q27Stop, qui conduirait à l’arrêt prématuré au bout du 27e acide aminé sur 121 de la protéine ORF8 du virus : il ne s’agit que d’une hypothèse, mais on pense que la perte de cette protéine n’aurait pas réellement de conséquences, et qu’ainsi l’arrêt prématuré de la production de cette protéine rendrait la réplication du virus plus efficace en évitant d’avoir à produire une protéine « non indispensable ».
A ce stade les informations suggèrent que cette souche est plus contagieuse, mais pas plus mortelle. Une étude – à prendre avec énormément de précautions – , de la London School of Hygiene and Tropical Medicine, estime que ce variant serait environ 56 % plus transmissible que les autres souches.
Le « variant Sud-Africain » ou B.135
Il possède un certain nombre de mutations dans la protéine Spike, dont la mutation N501Y. Mais il ne possède par exemple pas la délétion 69/70. Le ministre de la santé britannique, Matt Hancocksemble particulièrement préoccupé par ce variant. Il déclare lundi 4 janvier 2020 « c’est un problème vraiment, vraiment significatif, bien plus que le variant britannique ». Cela serait dû à cette accumulation de mutations dans la protéine Spike et à la crainte que cela ne permette à ce variant d’échapper à l’immunité générée par le vaccin Oxford-Astra Zeneca. Mais hormis le nombre important de mutations dans la protéine Spike, on a peu de données objectives à ce sujet, et il s’agit essentiellement de spéculations.
Le « variant Nigérian » ou B.1.1.207
On en sait à ce stade peu sur ce variant, si ce n’est qu’il possède la mutation P681H. On ne sait pas ou il aurait émergé, ni si ses mutations contribueraient à une sévérité ou mortalité accrue, ou même à une transmission accrue du virus, ce qui le distingue des deux autres « nouvelles souches ».
Il suffirait qu’un seul des nouveaux vaccins autorisés se montre nettement moins efficace contre une seule « nouvelle souche » pour que cela puisse avoir un impact considérable dans la confiance et l’adhésion envers ces nouveaux vaccins et ce à un moment critique de la campagne mondiale de vaccination contre le Sars-Cov-2.
Xavier Olessa-Daragon
8 – À quoi seraient dues ces accumulations de mutations ?
Il ne s’agit à ce stade que d’hypothèses, mais cela se serait produit « au sein » d’un patient immunodéficient infecté chroniquement par le virus. Chez ces patients, le génome viral se serait trouvé pendant 2 à 4 mois, parfois plus. Cela laisserait le temps au virus de se reproduire de façon extrêmement prolifique et d’accumuler énormément de mutations dans un premier temps. Le traitement que ces patients recevraient parfois tardivement, à base d’anticorps dérivés du plasma de patients guéris du virus, opérerait dans un second temps une « sélection » au sein du pool de diversité génétique créé dans un premier temps, des virus ayant notamment accumulé des mutations leur permettant de mieux résister au système immunitaire. Ces derniers pourraient enfin être transmis à d’autres patients. Ces évènements seraient extrêmement rares, mais tout comme le sont ceux qui ont donné naissance au Sars-Cov 2, il suffit d’un seul, suffisamment « efficace »… Quand on rappelle que les patients immunodéprimés, ne peuvent, pour la plupart pas être vaccinés pour des raisons évidentes, et que la prévention d’infection chez eux dépend d’une immunité collective à des taux qui sont aujourd’hui non seulement très loin d’être atteints, mais complexes à envisager pour des raisons qui dépassent la pure logistique, on continue de voir que l’apparition de ces nouvelles souches risque de peser lourd dans le débat sur la stratégie vaccinale.
9 – Où ont elle été trouvées jusqu’ici ?
La « souche britannique », B117, aurait été détectée dans au moins 33 pays, Australie, Belgique, Brésil, Canada, Chili, Chine, Danemark, Finlande, France, Allemagne, Islande, Inde, Irlande, Israël, Italie, Japon, Jordanie, Liban, Malte, Pays-Bas, Norvège, Pakistan, Portugal, Singapour, Corée du Sud, Espagne, Suède, Suisse, Taiwan, Turquie, Emirats Arabes Unis, Royaume-Uni et États Unis.
10 – Que surveiller ?
En plus du taux de mortalité de ces nouveaux variants qui arrivera comme toujours avec un certain délai, il faudra surveiller l’évolution des connaissances sur la contagiosité de ces nouveaux variants, mais également sur l’efficacité des vaccins. Il suffirait qu’un seul des nouveaux vaccins autorisés se montre nettement moins efficace contre une seule « nouvelle souche » pour que cela puisse avoir un impact considérable dans la confiance et l’adhésion envers ces nouveaux vaccins et ce à un moment critique de la campagne mondiale de vaccination contre le Sars-Cov-2. Enfin, s’il venait à être démontré qu’une de ces nouvelles souches largement répandue pouvait échapper à plusieurs vaccins, le problème pourrait n’être plus seulement politique mais devenir un véritable défi logistique supplémentaire. En effet quand bien même une « mise à jour » du vaccin dirigée contre une ou plusieurs de ces nouvelles souches pourrait être développée en quelques mois et de façon sûre, le défi logistique et politique n’en resterait pas moins considérable.
Enfin,en plus de ces nouvelles souches, il faut surveiller l’apparition de « nouvelles nouvelles souches » dérivées de ces « nouvelles souches » mais en y ajoutant encore de nouvelles mutations rendant le virus potentiellement capable d’échapper à l’immunité provoquée par les vaccins actuels, plus mortel, voir encore plus contagieux.
Une chose est sûre, plus le virus circule, plus le virus mute…
